Methylation
Proteins, one of the major classes of biological molecules perform a multitude of tasks within our cells and bodies. To make sure they do the correct job at the right time, they are very tightly regulated. One method of regulation is through the addition of chemical groups to the protein - methylation is one of these, and is able to change how a protein interacts with other molecules, when the protein is made and when the protein is destroyed - as well as in sensing changes in the environment (such as glucose). There is still a lot to find out about how this modification works - including what proteins are affected by it, how it is regulated, and what pathways it is active in. In my research, I want to explore the role of protein methylation in cancer, with the aim of identifying the underlying biology and potential new treatment opportunities.
Enrichment
We use a technique called mass spectrometry to identify and quantify methylated proteins. This approach is very powerful, allowing the discovery and quantification of many proteins simultaneously. However, one difficulty is in identifying methylated proteins (peptides) that are in very low abundance. One way to circumvent that is to "enrich" for these proteins prior to analysis. This can be very difficult and many techniques are available, but for proteins containing lysine methylations, the available approaches do not work. In collaboration with Chemists at Imperial College and the CRICK, we are developing novel chemical probes to target lysine methylation for enrichment. This will be a real "game-changer" in the field, allowing unparalleled access to methylated proteins.
Disease
Protein methylation is key regulatory process of many disease-associated proteins, including cancer (oncogenes) and neurological disease. Cancer is a disease in which the regulation of cell-cycling breaks down and cells proliferate with little or no control. In 15% of all cancers, a gene called MTAP is deleted. This gene is important for preventing the toxic build-up of a small compound, MTA, which is ultimately converted into SAM - the molecule that donates a methyl-group for protein methylation. This results in a reduction in the cellular abundance of the compound required for all protein methylation. In many cancers, this causes a vulnerability to processes requiring protein methylation and it has been shown that in these cases, cells are sensitive to inhibition of some methyltransferases (e.g. PRMT5). I am interested in exploring the relationship between metabolomic changes arising from MTAP deletion in cancer and protein methylation. I hope to uncover specific pathways that are sensitive to chemical inhibition when MTAP is deleted as this will provide us with new cancer-specific therapeutic opportunities.
Data analysis
Mass Spectrometry
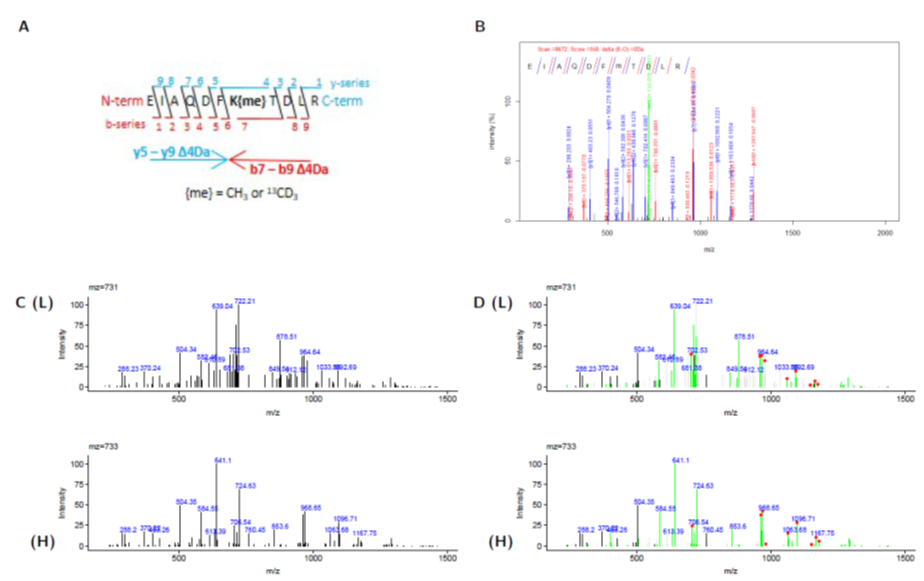
We use R to code our own bespoke data analysis tools for mass spectrometry data files. Recently, we published a tool, HiLight-PTM that highlights m/z peaks from peptide fragmentation (MS/MS) spectra that have matching masses, and those which have a predictable mass-shift owing to the incorporation of an isotopic label. Using this tool, we can extract pairs of MS/MS spectra that contain correlative fragments. Coupled to this, we use retention-time matching and compare the ratio of precursor isotopic peaks to match heavy/light peptide at both the MS1 and MS2 level. By doing this, we can interrogate when standard database searching fails (e.g. owing to poor fragmentation scans from co-isolation or low intensity). We also use experimental fragmentation matching to in silico fragment ions independant of precursor mass. In this way, we attempt to identify MS2 scans with unknown modifications or alterations to their primary sequence.
Data Analyis
Parenclitic Network Analysis
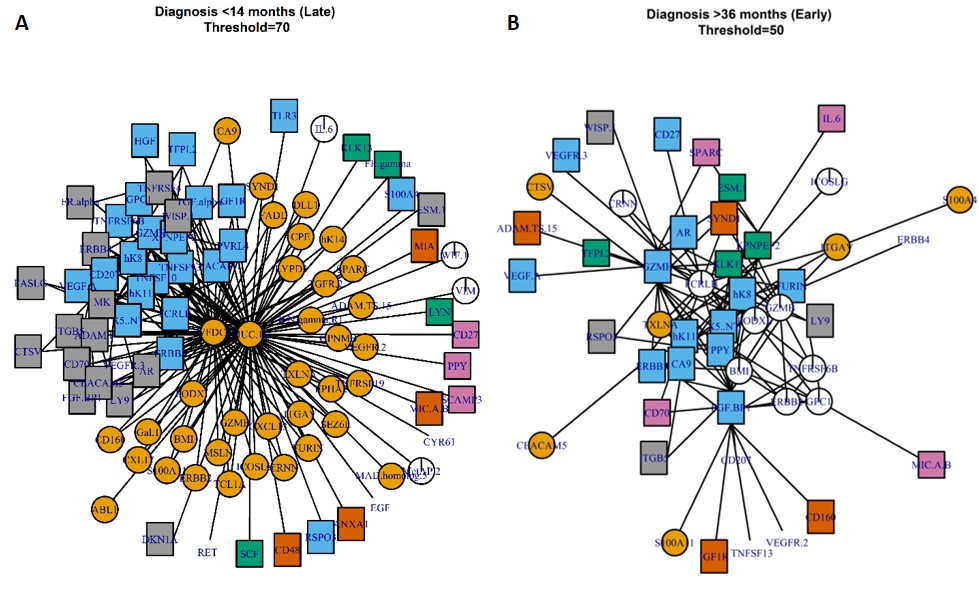
Parenclitic networks are networks of differences between two populations or between an individual and a population. First described by Zanin (2014), we have adopted them to better-suit biological data. The networks are constructed by performing comparisons of all pairs of analytes within in a dataset. A 2 dimensional kernel density estimatation is constructed for each pair in a control population and the density at which the same pair of analytes in a test population or individual lies within the control population is determined. If the density is above a threshold, then this relationship is plotted within a graph structure (network). With Prof Zaikin and Dr John Timms (UCL), we have used the topology (shape) of these networks to predict ovarian cancer and inform on early biological events in the pathogenesis of this disease. We have recently been awarded an MRC/NIHR methodological development grant to explore the applications of parenclitic networks and combine them with deep learning and machine learning approaches for disease prediction. We have a number of collaborations regarding the use of parenclitic networks (e.g. aging), and are happy to share code and explain their benefits further.
Data Analysis
Londitudinal Analysis
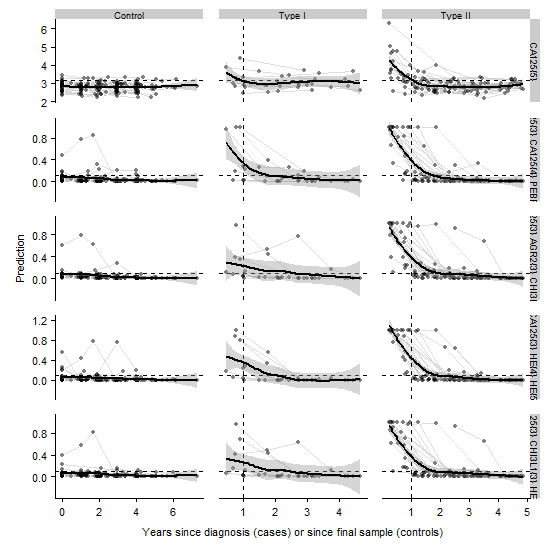
Standard biomarker tests use a fixed threshold at which a biomarker signifies disease. This threshold is calibrated against a population to give the optimal ratio of false positives to false negatives. Many biomarkers naturally vary in concentration in healthy individuals such that individuals with a naturally low abundance of a biomarkers may not be diagnosed if the marker doesn’t increase above threshold with the onset of disease (and vice-versa). Longitudinal algorithms that can use screening data are able to circumvent this problem by identifying changes in an individuals’ baseline (i.e. a rate of change). For example, we have developed a number of longitudinal algorithms able to identify ovarian cancer from screening data with improved sensitivity/specificity over threshold models. In collaboration with Dr Blyuss, I am exploring the application of these techniques in other fields, such as changes to protein expression over the cell cycle and longitudinal market data.